Автор: Денис Аветисян
Новое исследование показывает, как квантовые колебания протонов влияют на возбуждение электронов в органических соединениях, связанных водородными связями.
Купил акции по совету друга? А друг уже продал. Здесь мы учимся думать своей головой и читать отчётность, а не слушать советы.
Бесплатный телеграм-канал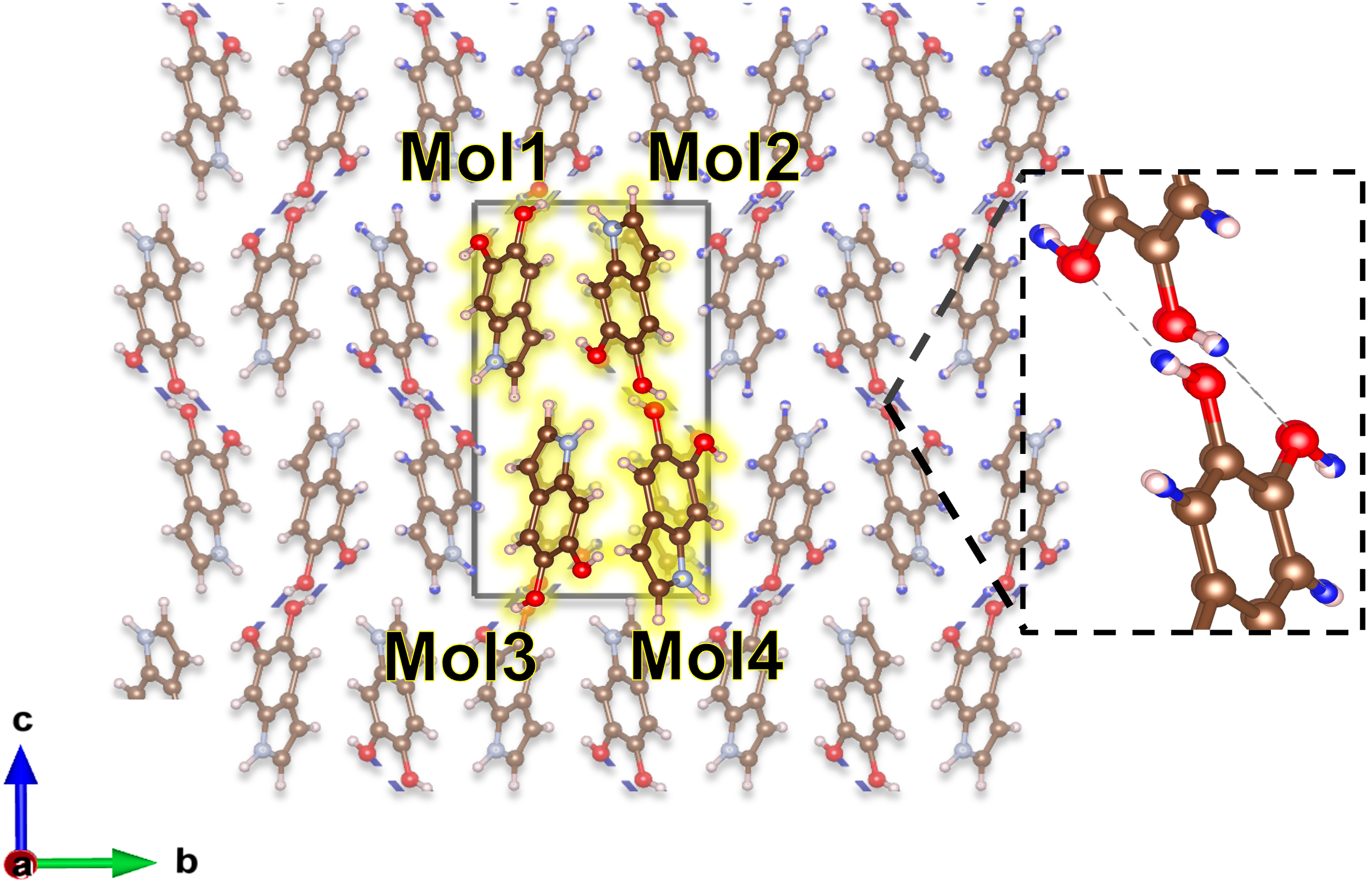
Применение методов NEO-DFT, G0W0 и уравнения Бете-Сальпетера позволило выявить влияние ядерных квантовых эффектов на распределение и анизотропию экситонов.
Несмотря на значительный прогресс в моделировании электронных свойств органических материалов, влияние квантовых эффектов протонов на процессы возбуждения электронов в системах с водородными связями остается малоизученным. В работе ‘Proton Quantum Effects on Electronic Excitation in Hydrogen-bonded Organic Solid: A First-Principles Green’s Function Theory Study’ представлен анализ влияния квантовых эффектов протонов на электронные возбуждения в органическом полупроводнике эумеланине, основанный на комбинации метода NEO-DFT, приближения G_0W_0 и уравнения Бете-Сальпетера. Показано, что квантование протонов приводит к заметным изменениям в распределении экситонов и формировании анизотропии на молекулярном уровне. В какой степени учет квантовых эффектов протонов может оптимизировать характеристики органических материалов для применения в оптоэлектронике и биофотонике?
Элегантность Эумеланина: Делокализация Экситонов и Квантовая Природа Связей
Эумеланин, распространенный пигмент, представляется уникальной системой для изучения делокализации экситонов, обусловленной его сложной структурой, формируемой водородными связями. В отличие от многих других органических молекул, эумеланин обладает разветвленной сетью этих связей, что создает сложный ландшафт потенциальной энергии для экситонов — возбужденных состояний, переносящих энергию. Данная сеть позволяет экситонам распространяться на значительные расстояния внутри молекулы, эффективно перенося энергию и влияя на оптические свойства пигмента. Изучение этой делокализации имеет важное значение для понимания механизмов защиты от ультрафиолетового излучения, осуществляемой эумеланином, а также для разработки новых материалов с контролируемыми оптическими характеристиками, вдохновленных природными системами.
Структура водородных связей в эумеланине играет определяющую роль в формировании его оптических свойств. Поскольку эумеланин поглощает и рассеивает свет уникальным образом, понимание расположения и силы этих связей позволяет предсказывать и даже контролировать его взаимодействие с электромагнитным излучением. Изменение конфигурации водородных связей, например, под воздействием температуры или химических факторов, непосредственно влияет на длину волны поглощаемого света и эффективность переноса энергии внутри молекулы. Таким образом, детальный анализ этих взаимодействий открывает возможности для создания новых материалов с заданными оптическими характеристиками, имитирующих или улучшающих естественные свойства эумеланина, что находит применение в разработке солнцезащитных средств, биосенсоров и оптических устройств.
Традиционные методы моделирования, применяемые для изучения водородных связей в эумеланине, часто оказываются недостаточными для полного описания квантово-механических нюансов этих взаимодействий. Суть проблемы заключается в том, что ядра атомов водорода не являются просто классическими частицами, а проявляют квантовые эффекты, такие как туннелирование и нулевые колебания. Эти эффекты оказывают значительное влияние на энергию и структуру водородных связей, а также на динамику переноса энергии в молекуле эумеланина. Игнорирование этих ядерных квантовых эффектов приводит к неточностям в расчетах оптических свойств пигмента и затрудняет предсказание его поведения в различных условиях. Более того, стандартные вычислительные подходы, основанные на классической механике, не способны адекватно описать корреляции между атомами водорода, что существенно ограничивает точность моделирования сложной водородной сети эумеланина.
Валидация Модели: Структура и Оптические Свойства
Рентгеноструктурный анализ был использован для определения спиральной структуры, формируемой водородными связями внутри эумеланина. Полученные данные о межмолекулярном расположении и параметрах спирали служат основой для последующего моделирования оптических свойств. Точное описание этой структуры необходимо для корректного расчета электронной структуры и, как следствие, спектра поглощения, поскольку взаимодействие между молекулами эумеланина существенно влияет на распределение электронов и оптическую активность материала. Информация о водородных связях, полученная из дифракционных данных, была интегрирована в вычислительную модель для обеспечения реалистичного представления межмолекулярных взаимодействий.
Для точного моделирования спектра оптического поглощения, структурные данные, полученные методом рентгеновской дифракции, были соединены с подходом NEO-MDFT (Near-Exact Optimized Effective Density Functional Theory). В расчетах использовался численный базис атомных орбиталей Tier 2, обеспечивающий оптимальный баланс между точностью и вычислительной эффективностью. Применение данного подхода позволило получить спектр поглощения, согласующийся с экспериментальными данными, что подтверждает адекватность выбранной методологии и параметров расчета для изучения оптических свойств эумеланина.
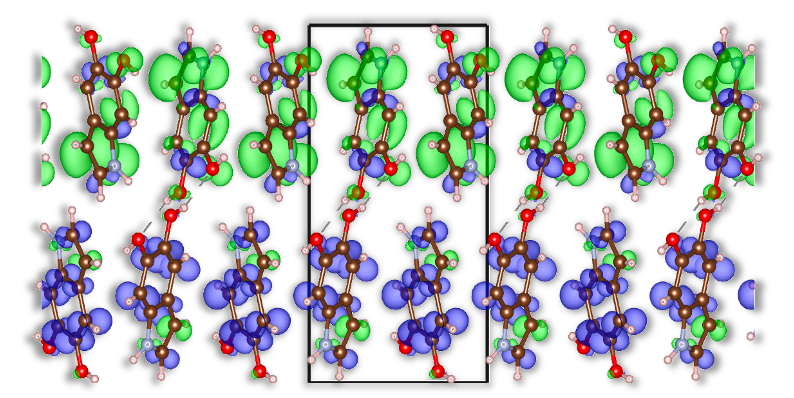
Анализ диэлектрической функции и плотности состояний, дополненный анализом распределения электронной плотности по методу Малликена, позволил получить важные данные о делокализации экситонов и оптическом отклике эумеланина. Проведенные расчеты показали, что учет квантовых эффектов протонов приводит к уменьшению ширины квазичастичной запрещенной зоны примерно на 0.05 эВ, а также к снижению энергии связи экситона с 1.46 эВ до 1.41 эВ. Полученные результаты свидетельствуют о существенном влиянии квантовых эффектов на оптические свойства материала.
Влияние на Дизайн Материалов и Перспективы
Точное моделирование ядерных квантовых эффектов, достигнутое, например, с помощью NEO-MDFT, является ключевым фактором в разработке материалов с заданными оптическими свойствами. Традиционные методы, рассматривающие атомы как классические частицы, часто оказываются неспособными адекватно описать поведение атомов водорода в сложных системах, таких как водородные связи. Влияние квантовых эффектов, таких как туннелирование протонов и нулевые колебания, может значительно изменять электронную структуру и, следовательно, оптические характеристики материала. NEO-MDFT, комбинируя преимущества методов теории функционала плотности с учетом ядерных квантовых эффектов, позволяет предсказывать и контролировать оптические свойства материалов на новом уровне точности, открывая возможности для создания инновационных оптических устройств и материалов с улучшенными характеристиками, например, для солнечной энергетики или биофотоники.
Исследование эумеланина, пигмента, отвечающего за окраску кожи, волос и глаз, выявило особенности делокализации экситонов - возбужденных состояний, участвующих в поглощении света. Установлено, что экситоны в эумеланине способны эффективно распространяться по молекулярной структуре, что обеспечивает высокую эффективность захвата света даже при слабом освещении. Данное понимание предоставляет основу для разработки биовдохновленных материалов с улучшенными характеристиками светопоглощения, которые могут быть использованы в солнечных батареях нового поколения, оптических сенсорах и других технологиях, требующих эффективного преобразования световой энергии. Имитируя структуру и свойства эумеланина, возможно создание искусственных систем, превосходящих существующие материалы по эффективности и устойчивости.
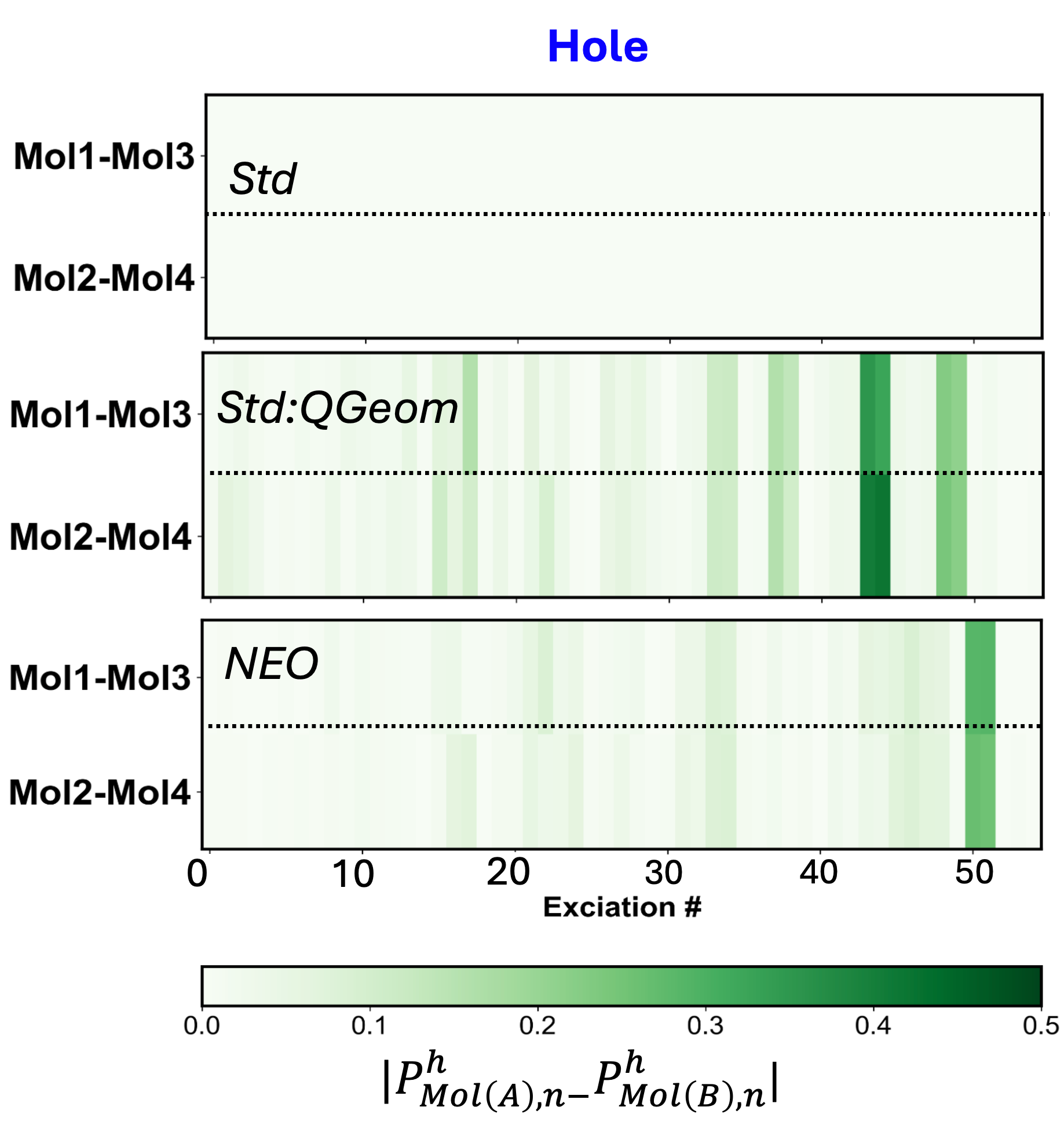
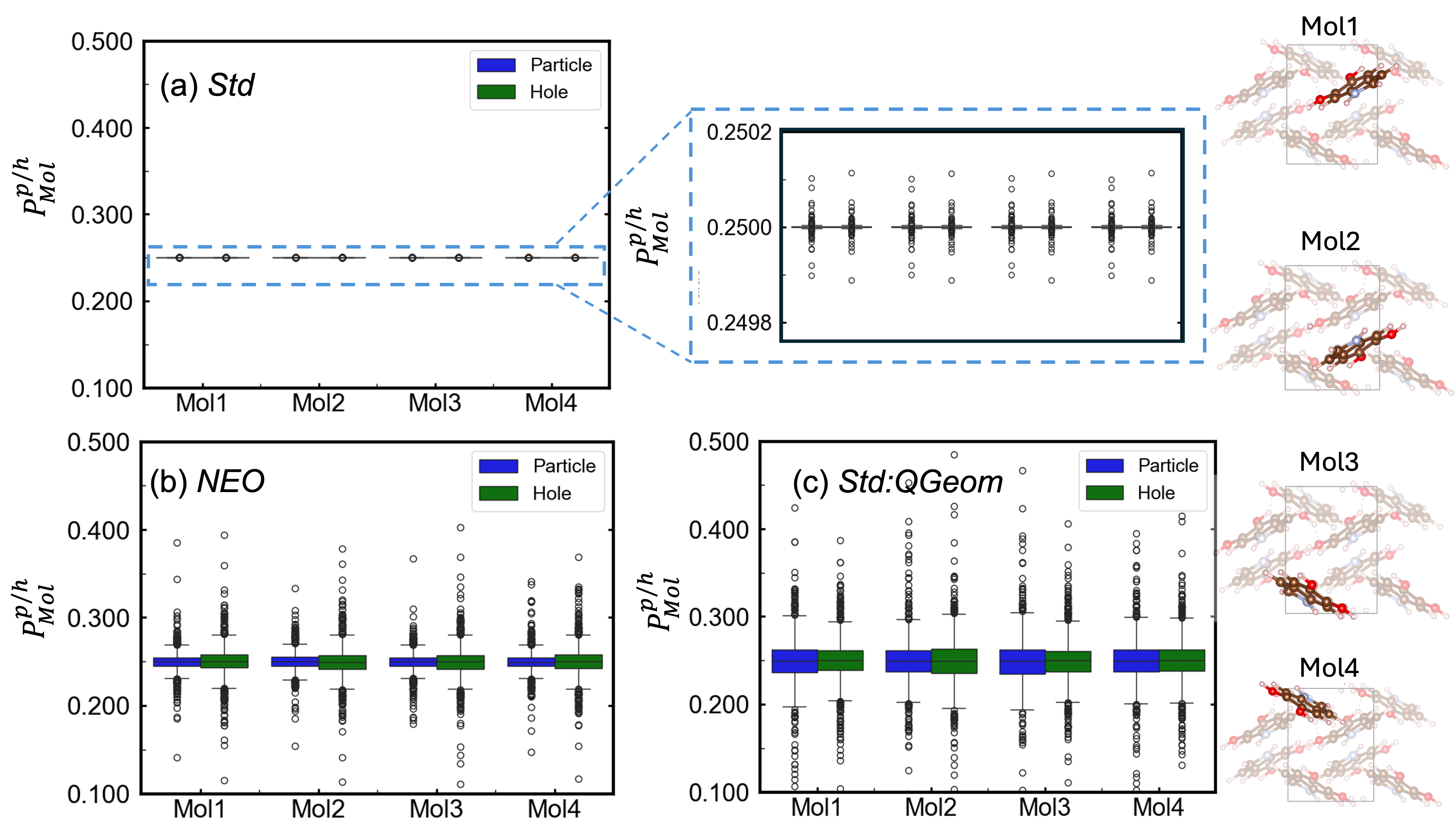
Разработанный объединенный теоретический и вычислительный подход открывает новые возможности для изучения связи между структурой и свойствами в широком спектре сложных систем, связанных водородными связями. Исследования показали, что учет квантования протонов приводит к увеличению стандартного отклонения популяций экситонов Мюллена в два порядка величины, что свидетельствует о значительном усилении анизотропии экситонов. Этот результат имеет важное значение для понимания и прогнозирования оптических свойств материалов, в особенности тех, где водородные связи играют ключевую роль в переносе энергии и формировании электронных состояний. Таким образом, предложенный метод позволяет более точно моделировать поведение сложных молекулярных систем и разрабатывать материалы с заданными оптическими характеристиками.
Исследование, представленное в данной работе, демонстрирует, как учет квантовых эффектов ядер, пусть N стремится к бесконечности - что останется устойчивым? - способен вносить заметные изменения в распределение и анизотропию экситонов в водородсвязанных органических твердых телах. Этот факт подчеркивает фундаментальную важность рассмотрения ядерных степеней свободы при моделировании электронных свойств материалов. Карл Саган однажды заметил: «Мы - звездная пыль, осознающая себя». Подобно тому, как звездная пыль формирует сложные структуры, так и учет квантовых эффектов позволяет более точно описывать сложное поведение материи на микроскопическом уровне, раскрывая истинную элегантность математической чистоты лежащих в ее основе принципов.
Куда Далее?
Представленная работа, хоть и демонстрирует влияние ядерных квантовых эффектов на электронные возбуждения в водородсвязанных органических твёрдых телах, не решает проблему фундаментальной непротиворечивости различных уровней теории. Использование NEO-DFT в сочетании с G0W0 и уравнением Бете-Сальпетера, безусловно, является шагом вперёд, однако, остаётся вопрос о систематической сходимости и предсказуемости. Достаточно ли этих приближений для описания сложных систем, или мы лишь приближаемся к истине, добавляя всё новые и новые параметры?
Очевидной задачей представляется расширение данной методологии на более крупные и сложные системы, содержащие большее количество водородных связей и взаимодействующих молекул. Необходимо учитывать влияние окружения, а также эффекты, связанные с динамическими искажениями кристаллической решетки. Простое увеличение вычислительных ресурсов не решит проблему, если базовая модель не способна адекватно описать физическую реальность.
В конечном счёте, истинная элегантность заключается не в сложности расчётов, а в простоте и ясности полученных результатов. И лишь тогда, когда теоретические предсказания будут согласованы с экспериментальными данными с высокой точностью, можно будет говорить о реальном прогрессе в понимании природы электронных возбуждений в конденсированных средах. Иначе это лишь игра чисел, лишённая всякого смысла.
Оригинал статьи: https://arxiv.org/pdf/2602.07791.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Лучшие шаблоны дивизий в Hearts Of Iron 4
- Решение головоломки с паролем Absolum в Yeldrim.
- Эпизод ‘Dungeons & Dealers’ Теда точно передает опыт D&D.
- Skyrim: 23 лучшие жены и как на них жениться
- Шоу 911: Кто такой Рико Прием? Объяснение трибьюта Grip
- Palworld: как получить ядра хищников
- Где посмотреть онлайн-фильм «Холодные ноги», ставший вирусным в TikTok
- Как пройти I’m Not a Robot – полное прохождение всех уровней
- Десять персонажей из следующего приквела ‘Йеллоустоуна’
- Лучшее оружие, броня и аксессуары, которые стоит получить в начале Crimson Desert.
2026-02-11 04:21