Автор: Денис Аветисян
Новый подход позволяет точно моделировать химические реакции, даже если они включают в себя элементы и катализаторы, не встречавшиеся в обучающей выборке.
Купил акции по совету друга? А друг уже продал. Здесь мы учимся думать своей головой и читать отчётность, а не слушать советы.
Бесплатный телеграм-канал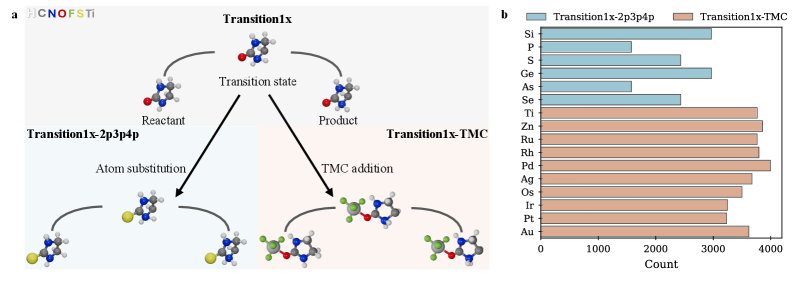
Исследователи разработали стратегию самообучения, использующую равновесные конформеры, для повышения обобщающей способности генеративных моделей в предсказании переходных состояний.
Несмотря на успехи в предсказании переходных состояний химических реакций, обобщение моделей машинного обучения за пределы тренировочного набора остается сложной задачей. В работе «Beyond the Training Domain: Robust Generative Transition State Models for Unseen Chemistry» представлены новые подходы к оценке и улучшению способности генеративных моделей предсказывать переходные состояния для реакций, включающих ранее не встречавшиеся химические элементы и комплексы переходных металлов. Предлагается стратегия самообучения, основанная на использовании равновесных конформеров, которая существенно повышает точность предсказания переходных состояний, снижая среднеквадратичное отклонение (RMSD) геометрии до 0.19 \mathring{A} для реакций T1x-TMC. Возможно ли создание универсального и масштабируемого фреймворка для изучения сложных реакционных ландшафтов, выходящих за рамки традиционной органической химии?
Вызов предсказания переходных состояний
Поиск переходных состояний в химических реакциях представляет собой серьезную вычислительную задачу, существенно ограничивающую возможности моделирования реакционной способности. Вычисление этих состояний, соответствующих точкам максимальной энергии вдоль реакционного пути, требует итеративных процессов оптимизации, которые становятся экспоненциально более затратными с увеличением числа атомов в системе и сложностью потенциальной энергетической поверхности. Вследствие этого, моделирование даже относительно простых реакций может потребовать значительных временных и вычислительных ресурсов, что препятствует исследованию сложных химических процессов, важных для таких областей, как разработка лекарств, материаловедение и катализ. Разработка более эффективных алгоритмов и методов для локализации переходных состояний является ключевой задачей современной вычислительной химии.
Традиционные методы поиска переходных состояний, такие как P-RFO (Principal Reaction Force Optimization) и IRC (Intrinsic Reaction Coordinate), зарекомендовали себя как надёжные инструменты в моделировании химических реакций. Однако, их применение сопряжено со значительными вычислительными затратами, особенно при исследовании сложных систем. Алгоритмы P-RFO требуют множества оптимизаций для нахождения наиболее вероятного пути реакции, а построение IRC — это ресурсоёмкий процесс, требующий последовательного вычисления энергии вдоль реакционной координаты. В случае систем с множеством степеней свободы или сложными потенциальными поверхностями, эти методы могут оказаться практически неприменимыми из-за экспоненциального роста необходимых вычислительных ресурсов и времени, что ограничивает возможность детального изучения кинетики и механизмов реакций в реальных условиях.
Машинное обучение для ускорения открытий
Генеративные модели, такие как React-OT и AEFM, представляют собой перспективный подход к быстрому предсказанию геометрий переходных состояний (ТС). В отличие от традиционных алгоритмов оптимизации, требующих итеративного поиска минимума энергии, эти модели обучаются на основе потенциальной энергетической поверхности и способны генерировать кандидаты в ТС более эффективно. Обучение происходит путем изучения корреляций между структурами молекул и их энергиями, что позволяет модели предсказывать геометрию ТС, минимизируя вычислительные затраты и время, необходимое для поиска. Это особенно важно при изучении сложных химических реакций, где поиск ТС является узким местом в расчетах.
Методы машинного обучения, такие как React-OT и AEFM, позволяют эффективно предсказывать геометрии переходных состояний, обучаясь на базовой поверхности потенциальной энергии (ППЭ). В отличие от традиционных алгоритмов оптимизации, требующих итеративного поиска минимумов энергии, эти модели способны генерировать кандидаты в переходные состояния значительно быстрее. Обучение на ППЭ позволяет им «понимать» закономерности в изменениях энергии при химических реакциях и, следовательно, предлагать более вероятные и точные геометрии переходных состояний, сокращая время вычислений и повышая эффективность поиска.
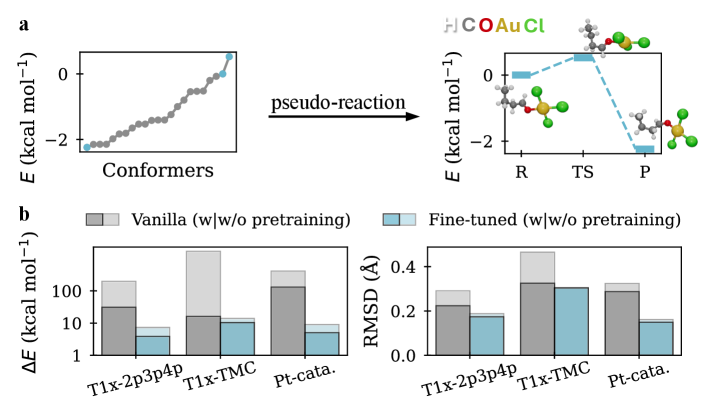
Предварительное обучение генеративных моделей псевдо-реакциями, полученными на основе структур равновесных конформеров, значительно повышает их эффективность в предсказании геометрий переходных состояний. В ходе исследований было установлено, что использование данной методики снижает медианное среднеквадратичное отклонение (RMSD) геометрий переходных состояний до 0.19 Å на бенчмарке Transition1x-TMC. Это указывает на существенное улучшение точности предсказаний по сравнению с другими методами, использующими только оптимизационные алгоритмы, и открывает возможности для ускорения процесса поиска переходных состояний в химических реакциях.
Оценка обобщающей способности и устойчивости
Для оценки обобщающей способности моделей, исходный набор данных Transition1x был расширен за счет добавления Transition1x-2p3p4p и Transition1x-TMC. Набор Transition1x-2p3p4p включает в себя реакции, содержащие элементы с формальными зарядами +2, +3 и +4, что значительно усложняет предсказание переходных состояний. Набор Transition1x-TMC представляет собой данные, полученные с использованием метода Transition State Theory with Multilevel Correction (TMC), который вносит дополнительные сложности, связанные с вычислением скоростей реакций и точностью определения переходных состояний. Включение этих расширенных наборов данных позволило более тщательно проверить способность моделей к экстраполяции и обработке разнообразных химических сценариев.
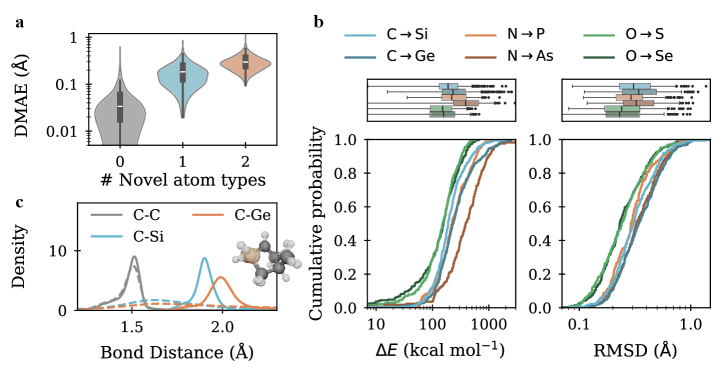
Для количественной оценки производительности моделей при предсказании переходных состояний использовались метрики, оценивающие геометрическое сходство и распределение длин связей между предсказанными и истинными структурами. Среднеквадратичное отклонение (RMSD) измеряет среднее расстояние между атомами в предсказанной и истинной структурах. Средняя абсолютная ошибка длины связи (DMAE) оценивает отклонение длин связей. Расстояние Вассерштейна-1 (Wasserstein-1 Distance) позволяет оценить разницу в распределении длин связей, что особенно важно для оценки качества предсказанных переходных состояний. Эти метрики в совокупности предоставляют комплексную оценку точности и надежности предсказаний.
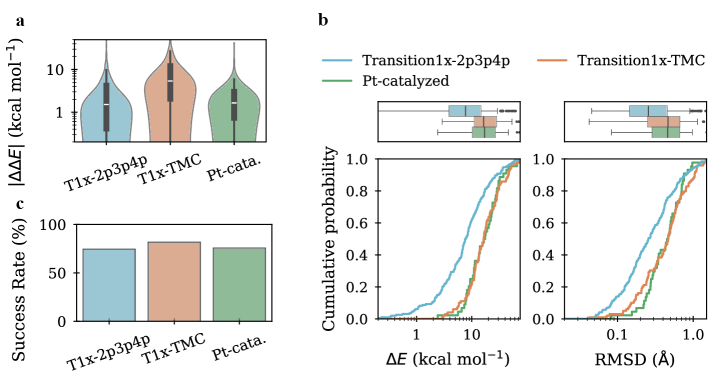
Модели показали значение среднеквадратичного отклонения (RMSD) в 0.26 Å на бенчмарке Transition1x-2p3p4p после реоптимизации сгенерированных образцов методами теории функционала плотности (DFT). На более сложном бенчмарке Transition1x-TMC было достигнуто улучшенное значение RMSD в 0.42 Å. Данные результаты демонстрируют значительное улучшение по сравнению с исходными показателями, что свидетельствует об эффективности предложенных методов в задаче предсказания переходных состояний.
Предварительное самообучение модели позволило снизить требования к объему данных для последующей донастройки на 75%. Это демонстрирует высокую эффективность обучения модели передаваемым представлениям, позволяющим эффективно адаптироваться к новым задачам при значительно меньшем объеме размеченных данных. Снижение потребности в данных достигается за счет предварительного изучения структуры данных без использования размеченных примеров, что позволяет модели сформировать базовое понимание, которое затем уточняется в процессе донастройки.
К будущему эффективного химического дизайна
Ускорение предсказания переходных состояний TS открывает новые возможности для высокопроизводительного скрининга условий реакций и кандидатов в катализаторы. Традиционно, определение TS являлось вычислительно затратным этапом в изучении химических процессов. Однако, современные методы машинного обучения позволяют значительно сократить время, необходимое для этого этапа, что, в свою очередь, даёт возможность исследовать огромное количество комбинаций условий и катализаторов. Этот подход особенно важен при поиске новых, более эффективных и селективных катализаторов, а также при оптимизации существующих химических процессов, позволяя находить оптимальные условия для достижения максимальной производительности и минимального образования побочных продуктов.
Значительное ускорение процессов химического открытия и оптимизации открывает новые горизонты в различных областях науки и техники. В фармацевтике, это позволяет быстрее разрабатывать и тестировать потенциальные лекарственные препараты, сокращая время и затраты на вывод новых препаратов на рынок. В материаловедении, возможность оперативно моделировать и предсказывать свойства новых материалов способствует созданию инновационных решений для энергетики, электроники и других отраслей. Такой подход позволяет исследователям эффективно исследовать огромные химические пространства, выявлять перспективные соединения и материалы с заданными характеристиками, тем самым значительно повышая эффективность научно-исследовательских работ и стимулируя технологический прогресс.
Сочетание методов машинного обучения с устоявшимися квантово-химическими расчетами, такими как DFT и GFN2-xTB, открывает перспективы для создания синергетичного подхода к эффективному и точному химическому моделированию. Данная комбинация позволяет использовать сильные стороны каждого метода: машинное обучение ускоряет предсказание переходных состояний и скрининг большого количества соединений, а квантово-химические расчеты обеспечивают высокую точность для наиболее перспективных кандидатов. Такой гибридный подход значительно снижает вычислительные затраты, позволяя исследовать химические реакции и свойства материалов с беспрецедентной скоростью и детализацией, что особенно важно для разработки новых лекарств и материалов с заданными характеристиками. В результате, исследователи получают возможность оптимизировать химические процессы и предсказывать свойства веществ с большей уверенностью, открывая новые горизонты в области химического дизайна.
Исследование демонстрирует стремление выйти за рамки привычных тренировочных данных, что особенно важно при моделировании реакций с участием новых химических элементов и сложных каталитических систем. Это созвучно взглядам Нильса Бора: “Противоположности не противоречат друг другу, а дополняют”. Подобно тому, как разные точки зрения обогащают понимание физических явлений, так и использование равновесных конформеров в качестве этапа предобучения позволяет генеративным моделям лучше обобщать предсказания переходных состояний, учитывая разнообразие химических пространств. Работа подчеркивает важность не только точного предсказания, но и способности модели адаптироваться к неизвестному, что является ключевым аспектом в развитии химического моделирования.
Куда двигаться дальше?
Представленная работа, безусловно, демонстрирует перспективность подхода, использующего самообучение на равновесных конформациях для повышения обобщающей способности генеративных моделей при предсказании переходных состояний. Однако, не стоит забывать старую истину: даже самая элегантная модель — лишь приближение к реальности. Воспроизводимость результатов на независимых наборах данных, включающих не только новые химические элементы, но и более сложные каталитические системы, остается критически важной проверкой. Если предсказания оказываются чувствительными к незначительным изменениям в параметрах модели или используемом программном обеспечении, то речь, скорее, о забавном трюке, а не о научном прорыве.
Особое внимание следует уделить оценке неопределенности предсказаний. Просто получить энергию переходного состояния недостаточно; необходимо понимать, насколько эта энергия надежна. Разработка методов, позволяющих количественно оценивать погрешность предсказаний, особенно в областях, где данные ограничены, представляется ключевой задачей. В конечном счете, ценность модели определяется не ее способностью генерировать красивые картинки, а ее способностью давать полезные предсказания с известной точностью.
Перспективы включают в себя интеграцию с другими методами вычислительной химии, такими как молекулярная динамика и квантовая механика. Создание гибридных подходов, сочетающих сильные стороны различных методов, может привести к более надежным и точным предсказаниям. И, конечно, не стоит забывать о необходимости постоянной проверки и совершенствования используемых алгоритмов, ведь наука — это не поиск абсолютной истины, а бесконечный процесс приближения к ней.
Оригинал статьи: https://arxiv.org/pdf/2601.16469.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Лучшее оружие, броня и аксессуары, которые стоит получить в начале Crimson Desert.
- Решение головоломки с паролем Absolum в Yeldrim.
- Шоу 911: Кто такой Рико Прием? Объяснение трибьюта Grip
- Лучшие шаблоны дивизий в Hearts Of Iron 4
- Все локации Тёмной Брони в Crimson Desert.
- Все коды в Poppy Playtime Глава 4
- Необходимо: Как выращивать урожай
- Все локации Святилищ в Crimson Desert
- Доллар обгонит вьетнамский донг? Эксперты раскрыли неожиданный сценарий
- Разработчики Call of Duty считают повышение цены на Game Pass ‘позитивным’.
2026-01-26 22:57